



Aliment Pharmacol Ther. 2016;44(9):946-956.
Abstract and Introduction
Abstract
Background Virologic and safety outcomes of ombitasvir/paritaprevir/ritonavir ± dasabuvir ± ribavirin (OBV/PTV/r ± DSV ± RBV) therapy have shown high sustained virologic response (SVR) rates and good tolerability in most patient populations in pre-registration studies.
Aim To confirm these clinical trial findings in the treatment of genotype 1 and 4 hepatitis C under real-world conditions.
Methods Patients enrolled for treatment with OBV/PTV/r ± DSV ± RBV based on therapeutic guidelines were included, and the regimen was administered according to product characteristics. Clinical and laboratory data, including virologic response, were collected at baseline, end of treatment (EOT) and 12 weeks after EOT.
Results A total of 209 patients with chronic hepatitis C were enrolled, most were genotype 1b-infected (84.2%) and 119 (56.9%) had liver cirrhosis. Among these, 150 (71.7%) had failed previous anti-viral therapies and 84 (40.2%) were null-responders. At 12 weeks after EOT, SVR was achieved by 207 (99.0%) patients, ranging from 96.4% to 100.0% across subgroups. All Child–Pugh B and post-orthotopic liver transplantation patients achieved SVR. Adverse events occurred in 151 (72.2%) patients and were mostly mild and associated with the use of RBV. Serious adverse events, including hepatic decompensation, renal insufficiency, anaemia, hepatotoxicity and diarrhoea, were reported in eight (3.8%) patients. In five (2.4%) patients, adverse events led to treatment discontinuation. On-treatment decompensation was experienced by seven (3.3%) patients.
Conclusions The results of our study confirm previous findings. They demonstrate excellent effectiveness and a good safety profile of OBV/PTV/r± DSV±RBV in HCV genotype 1-infected patients treated in the real-world setting.
Introduction
Hepatitis C virus (HCV) infection is the leading cause of chronic liver disease, affecting over 100 million people worldwide.[1] Approximately 20–30% of patients with chronic HCV infection develop liver cirrhosis within two to three decades from disease onset.[2] Chronic HCV infection is also associated with an increased risk of hepatocellular carcinoma (HCC) and contributes to one-third of HCC cases globally.[3]
The goal of treatment in patients with chronic HCV infection is HCV ribonucleic acid (HCV RNA) clearance from serum.[4] Sustained virologic response (SVR) is demonstrated by HCV RNA negativity at a specific time point after the end of treatment (EOT). Achievement of SVR reduces the risk of liver complications and translates into definitive virologic cure in more than 99% of patients.[4] Until recently, the standard of care in most HCV-infected individuals, and the only viable option, was treatment with a combination of pegylated interferon alpha (PEG-IFN) and ribavirin (RBV). The regimen provided SVR in 41–73% of patients, with significantly lower rates observed in genotype 1, the most common HCV subtype.[5] In addition to limited efficacy, PEG-IFN-based therapies were handicapped by numerous side effects that frequently resulted in premature treatment termination. Although the introduction of protease inhibitors (PIs; boceprevir and telaprevir) improved therapeutic outcomes in patients with HCV genotype 1, the regimen was ineffective in individuals infected with other HCV genotypes, prior null-responders to interferon-based therapies, and in those with liver cirrhosis.[6–8]
Development of second-generation direct-acting anti-virals (DAA) marked the advent of new anti-HCV therapies characterised by excellent SVR rates and a good safety profile, independent of liver fibrosis and previous treatment history.[9,10] Integrating DAA with different mechanisms of action ensured better virologic response and provided a robust genetic barrier to the emergence of drug-resistant HCV variants. The combination of the NS5A inhibitor ombitasvir (OBV), the NS3/4A PI paritaprevir (PTV) boosted with ritonavir (r) and the NS5B polymerase inhibitor dasabuvir (DSV), with or without RBV, was the first all oral interferon-free regimen containing three distinct DAAs approved for therapy of patients infected with HCV genotype 1 and 4, who are both treatment-naïve and treatment-experienced.[11–13] Although the combination proved effective and well tolerated, these were all clinical trials with stringent inclusion criteria and treatment conditions that are substantially different from those observed in normal medical practice.
Scarcity of data regarding the use of novel HCV therapies outside of the clinical trial setting creates the demand for real-life population-based analyses. To address this need, the AMBER multicentre, open-label, investigator-initiated study was conducted, with the aim to investigate the effectiveness and safety of OBV/PTV/r ± DSV ± RBV under 'real-world' conditions.
Materials and Methods
This study involved the collection of data from 16 hepatologic centres in Poland. Anti-viral medication was obtained within a named patient programme created to provide early access to patients with advanced liver disease and a life-threatening course of HCV infection. Since the treatment was based on regimens not approved in the European Union at the time of enrolment, each participating centre obtained approval for the experimental therapy and the study protocol from a local ethics committee. The study was conducted with respect to Good Clinical Practice guidelines and the Declaration of Helsinki principles for ethical research. Written informed consent was obtained from the participants, and all patient data were de-identified.
Study Population
Eligible patients were adults, male or female, with chronic HCV genotype 1 or 4 infection, previously treated or treatment-naïve, with liver cirrhosis, or post-orthotopic liver transplantation (OLTx). Patients with noncirrhotic livers were also included if an intolerance to PEG-IFN, treatment-limiting adverse events associated with prior PEG-IFN use, contraindications to PEG-IFN-based regimen, clinically significant extrahepatic manifestations of HCV infection or rapidly progressive fibrosis (according to the judgment of treating physician) were documented. Individuals co-infected with hepatitis B virus, with a life expectancy of <1 year, requiring haemodialysis or peritoneal dialysis, presenting with a neoplasm other than HCC that would require systemic chemotherapy during DAA treatment, diagnosed with new or recurrent HCC within the past 12 months, who had OLTx followed by ineffectively treated hepatic vasculature or biliary duct complications, with creatinine clearance <30 mL/min, haemoglobin <10.0 g/dL, or platelet counts <25 × 103/μL were excluded from the study. Patients were enrolled for treatment with OBV/PTV/r ± DSV ± RBV according to the therapeutic guidelines of the HCV Polish Expert Group and the manufacturer's recommendations.[14]
Medication and Follow-up
The dose of OBV/PTV/r (Viekirax 12.5 mg/75 mg/50 mg; AbbVie Deutschland GmbH & Co, Ludwigshafen, Germany) was 25 mg/150 mg/100 mg/day and DSV (Exviera 250 mg, AbbVie Deutschland GmbH & Co, Ludwigshafen, Germany) was 500 mg/day divided in two doses. The dose of RBV was 1000 mg/day in patients weighing <75 kg or 1200 mg/day in those weighing >75 kg. Individuals presenting with significant laboratory abnormalities at baseline (anaemia, thrombocytopenia, impaired renal function) were started on RBV with dose adjusted according to product characteristics. RBV dose was modified or discontinued during therapy in patients who developed severe adverse events or laboratory abnormalities. The addition of RBV was required in all patients except those with noncirrhotic livers infected with genotype 1b. DSV was not administered to HCV genotype 4-infected patients. Treatment duration was 12 weeks in the majority of patients, but was extended to 24 weeks in patients with liver cirrhosis infected with HCV genotype 1a, 1 (if subgenotyping was not available), or 4, and in all post-OLTx patients.
Patients were assessed at day 0 (baseline), EOT and 12 weeks after the EOT (FU12). However, at the discretion of the treating physician, additional visits were also possible at day 1, day 7, week 4 and week 8. Baseline clinical data included gender, age, body weight, fibrosis status according to METAVIR score, treatment history and comorbidities. Fibrosis was evaluated based on the available reports from liver biopsy or elastography carried out with either transient elastography or shear-wave elastography or acoustic radiation force impulse. Individuals who failed previous PEG-IFN-based therapies were categorised as null-responders, partial responders or relapsers. The remaining proportion was treatment-naïve, or their treatment history was unknown. Laboratory data included HCV RNA level, liver function tests (bilirubin, alanine transaminase – ALT, alkaline phosphatase, international normalised ratio, INR and albumin), platelets count, haemoglobin concentration (Hgb), alpha fetoprotein and serum creatinine. Child–Pugh and Model for End-Stage Liver Disease (MELD) scores were calculated at baseline, EOT and FU12.
Virologic and Safety Assessment
The primary effectiveness endpoint was the proportion of patients with SVR12 (undetectable HCV RNA at FU12). HCV RNA detection level was <18 IU/mL but varied across study centres depending on the assay used (<15 IU/mL in 88.0% of patients). Secondary endpoints were to assess the usefulness of interim viral response in forecasting therapeutic outcomes and identify potential predictors of rapid virologic response (RVR; undetectable HCV RNA at 4 weeks) that could justify possible shortening of treatment duration. HCV RNA levels were obtained by quantitative PCR assays: COBAS TaqMan HCV v2.0 (Roche Molecular Diagnostics, Pleasanton, CA, USA), COBAS AmpliPrep HCV (Roche Molecular Diagnostics, Pleasanton, CA, USA) and m2000 RealTime System (Abbott Molecular, Des Plaines, IL, USA). Samples were analysed according to the intention-to-treat principle.
The primary safety endpoints were adverse events reported by investigators from baseline to 30 days following the final dose of the study medication. The secondary safety endpoint was to investigate possible predictors of on-treatment hepatic decompensation, defined as development of hepatic encephalopathy or ascites in patients without these signs at the initiation of treatment.
Statistical Analysis
Data are presented as mean ± standard deviation (s.d.) unless indicated otherwise. Serum HCV RNA levels were logarithmically transformed. Mann–Whitney U and Kruskal–Wallis anova tests were used for univariate and multivariate comparisons of independent continuous variables; Fisher's exact test was applied to compare discrete variables. Multivariate analyses were performed using a general regression model with undetectable HCV RNA at 4 weeks as a dependent factor. A 0.05 significance level was assumed for all tests. For the evaluation of predictive value and determining the optimal cut-off for selected markers, area under the receiver operating characteristic curve (AUROC) was performed. Statistical analyses were performed with statistica 12.0 (Statsoft, Tulsa, OK, USA) and GraphPad Prism 5.0 (GraphPad Software, Inc, La Jolla, CA, USA).
Results
Study Population
The demographical and clinical baseline characteristics are shown in Table 1. A total of 209 patients with chronic HCV genotype 1 (mostly 1b) and four infection were enrolled, including 150 (71.8.0%) nonresponders to prior PEG-IFN + RBV therapy, including 84 (40.2%) null-responders. A total of 16 nonresponders had failed previous triple regimens (PEG-IFN + RBV + PI). Liver cirrhosis was diagnosed in 119 (56.9%) patients, and 56 (26.8%) patients in this group were also null-responders. A total of 21 (10.0%) patients had a history of OLTx, and 14 (6.7%) were classified as Child–Pugh B prior to the initiation of treatment. Seven patients discontinued treatment but were still followed up and had their serum tested for HCV RNA at 12 weeks after treatment discontinuation. Patient disposition, including reasons for treatment discontinuation, is shown in Figure 1.
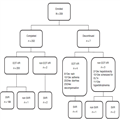
Figure 1.
Patient disposition diagram including reasons for discontinuation. EOT-VR, end of treatment virologic response; OLTx, orthotopic liver transplantation; SVR, sustained virologic response.
Effectiveness
The majority (207/209, 99.0%) of patients achieved SVR12 with rates ranging from 96.4% to 100.0% across subgroups (Figure 2). The only two nonresponders were patients infected with the HCV genotype 1b, with liver cirrhosis, who were null-responders to prior PEG-IFN + RBV therapy. Among the 202 patients who completed the scheduled therapy, two (1.0%) patients were HCV RNA positive at EOT but had viral load undetectable at FU12. Of the two patients who failed to achieve SVR12, one relapsed after discontinuation at week 2 due to suspicion of grade 3 hepatotoxicity (ALT 780 IU/mL, bilirubin 8 mg/dL), and one had a post-treatment relapse despite undetectable HCV RNA at the EOT. Baseline factors associated with higher effectiveness were as follows: male sex, HCV genotype 1a or 4 infection, absence of cirrhosis, no history of previous anti-viral treatment, HCV RNA <700 000 IU/mL, platelets count ≥100 000 cells/μL and serum albumin ≥3.5 g/dL. The differences were, however, of no significance, and effectiveness exceeded 96% in all subgroups analysed according to baseline factors (Figure 3).

Figure 2.
SVR12 rates in general patient population and across subgroups. OLTx, orthotopic liver transplantation; PEG-IFN, pegylated interferon; PI, protease inhibitor; RBV, ribavirin; SVR12, sustained virologic response at 12 weeks after the end of treatment.

Figure 3.
SVR12 rates across according to baseline factors (%). SVR12, sustained virologic response; PLT, platelet count.
In the 59 patients with virologic data available, viral load declined by 1–4 logs after 1 day, with 10 (16.9%) patients demonstrating undetectable HCV RNA (Figure 4). Undetectable viral load after 1 week was demonstrated in 27 of 84 available (32.1%) patients and had a 100% positive predictive value for the achievement of RVR. RVR was achieved in 110 of 186 available (59.5%) patients and was associated with baseline HCV RNA level (P = 0.003), MELD (P = 0.01), Child–Pugh score (P = 0.02) and the diagnosis of arterial hypertension (P < 0.001) in a univariate analysis. Multivariate analysis showed that low viral load with a cut-off of 700 000 IU/mL and a Child–Pugh score <6 at baseline were independent factors contributing to undetectable HCV RNA at 4 weeks (P = 0.03 and P = 0.021, respectively). Baseline HCV RNA level had high predictive value for the achievement of RVR in the ROC analysis (AUC = 0.63, P = 0.04). Presence of liver cirrhosis, tissue stiffness on elastography, RBV use, type of previous nonresponse or laboratory measures (bilirubin, albumins, INR, ALT and platelet count) were not associated with RVR when analysed separately. The highest week 4 response rate of 76% was demonstrated in patients with baseline HCV RNA level <700 000 IU/mL and Child–Pugh score <6, whereas combination of higher HCV RNA levels and Child–Pugh scores provided RVR in only 17% of patients.
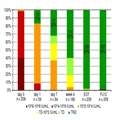
Figure 4.
HCV RNA dynamics during treatment and follow-up. EOT, end of treatment; FU12, follow-up at 12 weeks after the end of treatment; TD, HCV RNA detectable but below the limit of quantification; TND, HCV RNA undetectable.
Safety
No deaths occurred during the treatment and the 12-week follow-up period. One patient died due to carcinoma in the bones without liver involvement (primary lesion unknown), which was diagnosed 5 months after treatment termination. Adverse events (mostly asthenia, fatigue, nausea and headache) were reported in 151 (72.2%) patients, more frequently in those treated with RBV (Table 2). Serious adverse events were experienced by eight (3.8%) patients, who were all treated with RBV, and included hepatic decompensation, anaemia, diarrhoea, hepatotoxicity and renal insufficiency. The latter developed in a patient with a history of OLTx and was related to tacrolimus despite dose reduction according to the label. Treatment was discontinued in six patients, and was related to adverse events in five (2.4%) patients. On-treatment hepatic decompensation occurred in seven (3.3%) patients: in four patients manifesting as ascites, in two patients as encephalopathy and in one patient as both. Of the patients who developed decompensation, one required OLTx after treatment termination, two were listed for OLTx, and in the remaining individuals, the episode resolved without a need for transplantation. According to treating physicians in two patients, decompensation was possibly associated with ongoing anti-viral therapy, while in the remaining patients, decompensation was considered to be disease-related. In all cases except one, the treatment was continued with special caution for possible progression of hepatic deterioration. During the treatment period one case of HCC was identified in a patient that had originally showed no lesion in the liver on ultrasound examination at baseline. Within at least 6 months of the post-treatment follow-up (range 7–16 months; mean 12.3 ± 2.2 months), HCC was diagnosed in one patient (1 month after treatment termination); this was a recurrence of HCC that had been diagnosed and treated with OLTx 2 years prior to the current HCV treatment. Decline in haemoglobin level below 10 g/dL was observed more frequently in patients treated with RBV (9.6% vs. 3.8%), whereas values below 8 g/dL were found at similar rates (~2.0%). On-treatment anaemia required RBV dose reduction in 33 (21.2%) patients, RBV discontinuation in eight (5.1%) patients, and blood transfusion in two (1.3%) patients treated with RBV. Total bilirubin level greater than three times the upper limit of normal (ULN) was only observed in patients receiving RBV (17.9%).
Predictors of Hepatic Decompensation
All seven patients who developed on-treatment hepatic decompensation were infected with genotype 1b and were diagnosed with liver cirrhosis. Six of these patients had a history of decompensation, and three patients were post-OLTx (Table 3). In decompensated patients compared with those without decompensation, baseline bilirubin (2.4 ± 1.3 vs. 1.1 ± 0.7, P = 0.001), INR (1.3 ± 0.2 vs. 1.1 ± 0.2, P = 0.003), albumin (3.3 ± 0.4 vs. 4.2 ± 2.6, P < 0.001), as well as the Child–Pugh (6.9 ± 1.1 vs. 5.3 ± 0.7, P < 0.001) and MELD scores (13.0 ± 2.0 vs. 8.2 ± 2.5, P < 0.001) were significantly different. The ROC analysis showed high discriminatory power to predict on-treatment decompensation for albumins (AUC = 0.88, P < 0.001), bilirubin (AUC = 0.84, P = 0.001) and INR (0.81, P = 0.005), with a likelihood ratio of 5.7 for <3.6 g/dL, 5.8 for >1.65 mg/dL and 6.2 for >1.28, respectively. All decompensated patients had received RBV. On-treatment ALT elevations >2 times the ULN were not observed in patients who developed hepatic decompensation, so we can assume that they were not associated with hepatotoxicity.
Concomitant Medications
Concomitant medications were used by 140 (67.0%) patients, with 33 (15.8%) taking ≥5 different drugs. The major therapeutic groups were cardiovascular and anti-hypertensive medications (62 patients), diuretics (41 patients), gastrointestinal agents (32 patients), immunosuppressants (29 patients) and hypoglycemics (21 patients). Prior to the initiation of anti-viral therapy, concomitant medications were discontinued or replaced in 10 (4.8%) patients and dosage was modified in 29 (13.9%) in order to avoid drug–drug interactions; this most frequently pertained to tacrolimus (11 patients), ciclosporin (5 patients), amlodipine (5 patients) and furosemide (3 patients).
Discussion
This study provides real-world evidence for excellent anti-viral potency of OBV/PTV/r ± DSV ± RBV in the treatment of HCV genotype 1 and 4 infection. Virologic success was achieved by 99% (207/209) of patients, with SVR12 rates ranging from 96.4% to 100% across subgroups. Virologic failure was only experienced by two patients (1%), who were both infected with the HCV genotype 1b, had liver cirrhosis and were null-responders to previous PEG-IFN-based regimens. Our results confirm the similar low rates of post-treatment relapse in individuals treated with OBV/PTV/r ± DSV ± RB previously observed in clinical trials and emerging real-world data.[11,15]
The regimen also proved highly effective in difficult-to-treat populations, such as those with liver cirrhosis or null-responders to previous anti-viral therapies. SVR12 was achieved in 98.3% (117/119) of patients with liver cirrhosis irrespective of their treatment history, in agreement with the results of previous clinical trials and real-world studies on patients with cirrhosis (85.9–100%).[13,16–19] The SVR12 rate in nonresponders to previous therapies reached 98.7% (148/150) similar to that observed in pre-registration trials (92–100%).[13,16,18–22] SVR12 was achieved in 97.6% (82/84) of null-responders to prior PEG-IFN + RBV therapy, irrespective of the presence of cirrhosis, confirming the rates (85.9–100%) of prior registration studies in patients without cirrhosis or with compensated cirrhosis.[13,16,18–22] The only clinical trial to investigate outcomes of OBV/PTV/r ± DSV ± RBV therapy in individuals with a history of hepatic decompensation was performed in a group of 11 Child–Pugh B patients, who all achieved SVR12.[23] These findings were replicated in the real-world conditions of the AMBER study (SVR12 100%, 14/14). Moreover, the SVR12 rate was 100% in liver transplant recipients (21/21), consistent with the results of the CORAL study that demonstrated an SVR12 rate of 96–100% in post-OLTx patients.[24] Finally, while data regarding virologic outcomes in individuals who failed PEG-IFN + RBV + PI 'triple' therapy is limited, we observed 100% effectiveness (16/16 patients) in this group. A similar SVR rate of 96.8% was recently demonstrated in a population of 62 patients who failed previous boceprevir- or telaprevir-based regimen.[25]
OBV/PTV/r ± DSV ± RBV treatment proved generally safe and well tolerated. While adverse events occurred in 72.2% of patients, they were mostly mild or moderate, including asthenia, fatigue, nausea and headache. Similar observations come from registration studies, with adverse events reported in 74% of subjects in safety reports from phase 2 and 3 clinical trials.[26] Serious adverse events were also infrequent (3.8% of subjects). We found grade 3 and 4 ALT elevation was related to RBV administration in our study, which is consistent with pooled safety data from phase 2 and 3 clinical trials that demonstrated higher rates of ALT elevation in patients treated with RBV.[26] Furthermore, hepatic decompensation during OBV/PTV/r ± DSV ± RBV therapy was observed in seven patients, although six of these patients had a history of decompensation, a crucial risk factor for decompensation during anti-HCV therapy. Since there was only one case of HCC in the post-treatment follow-up (<0.5%), our data do not support enhanced risk of HCC development after DAA-based regimen.[27]
Although OBV/PTV/r ± DSV ± RBV is currently not recommended in patients with Child–Pugh B liver disease following post-marketing reports of hepatic decompensation and failure, the causal relationship between its use and the exacerbation of pre-existing liver function impairment has not been established.[28–30] Child–Pugh B patients were enrolled in the AMBER study before these recommendations were released by the European Medicines Agency. All Child–Pugh B patients including those who developed on-treatment decompensation achieved SVR12. Our findings suggest that individuals with moderate liver disease may still benefit from the treatment; however, in cases with a history of hepatic decompensation or current signs of hepatic functional impairment, other therapeutic options should be considered.
Although most patients in the AMBER study took at least one concomitant medication, pharmaceutical interactions were only expected in 15.8% of cases. In all cases, treatment was modified or discontinued prior to the initiation of anti-viral therapy, as recommended in the product characteristics.[27,28] Therefore, our real-world data indicate that, despite potential drug–drug interactions identified in phase 1 and 2a clinical trials, the use of concurrent medication may not be problematic and should not discourage the decision to treat.
This study has several limitations. First, the method of assessment of liver fibrosis varied across patients (i.e., liver biopsy and several non-invasive techniques were used), which could have influenced the results. While cirrhosis status was evaluated not earlier than 1 year before the initiation of treatment, the time of the evaluation differed among study individuals. This variation may have underestimated the prevalence of cirrhosis, especially in cases of rapidly progressing disease. Second, an open-label study design inherent to all real-world observations may have biased safety outcomes through underreporting of the adverse events, a phenomenon known as 'tendency to please'. Last, the numbers of patients in some of the subgroups were relatively low. For example, although treatment with OBV/PTV/r ± DSV ± RBV showed 100% effectiveness in patients infected with genotype 4, we must keep in mind that only nine patients infected with this genotype were enrolled. This sample size is too small to make any definitive conclusions.
In summary, OBV/PTV/r ± DSV ± RBV therapy proved highly effective in patients with chronic HCV infection, including individuals with advanced liver disease, a history of nonresponse to PEG-IFN-based regimens, or post-OLTx. Adverse events were mostly mild and rarely led to treatment discontinuation. Episodes of ascites or encephalopathy were infrequent and occurred in patients with cirrhosis, most of whom had a history of hepatic decompensation. Therefore, given its outstanding virologic outcomes and low risk of serious adverse events, the OBV/PTV/r ± DSV ± RBV regimen is a good treatment option for patients with chronic HCV genotype 1 infection.